Was sind die molekularen Ursachen der Spinalen Muskelatrophie ?
Spinale Muskelatrophie (SMA) ist eine genetische Krankheit die ca. eine Person unter 6.000 betrifft. Charakteristisch bei dieser Erkrankung ist das Absterben der Motorneurone, also der Zellen, die unseren Muskeln Signale geben, sich zusammen zu ziehen („kontrahieren“). Dies erklärt auch das Krankheitsbild: SMA-Patienten entwickeln im Laufe ihres Lebens eine fortschreitende Lähmung der Gliedmaßen und des Rumpfes.
Was bewirkt das Absterben von Motorneuronen im Rückenmark von SMA-Patienten und wie kann man das verhindern? Dies ist eine der wichtigen Fragen, mit der sich SMA-Forscher aus unterschiedlichen Disziplinen, wie der Medizin, Genetik, Neurobiologie und Biochemie seit einigen Jahren intensiv beschäftigen. Als erster Schritt zum “Ziel” (d.h. einer Therapie der Krankheit) wurde 1995 das Krankheitsgen durch Judith Melki identifiziert. Es handelt sich um das survival motor neuron (SMN) Gen, welches für ein Protein codiert, das in Patienten mit SMA nicht mehr in ausreichenden Mengen gebildet wird. Wie sich durch biochemische und zellbiologische Studien schnell herausstellte, handelt es sich um ein Protein mit bemerkenswerten z. T. zuvor noch völlig unbekannten Eigenschaften. Zunächst war man überrascht, dass das SMN-Protein nicht nur in Motoneuronen, d.h. den zuerst betroffenen Zellen bei SMA-Patienten, sondern in nahezu allen Zellen des Körpers gleichermaßen gebildet (“exprimiert”) wird. Es ist daher, wie die Biochemiker sagen, ein “Haushaltsgen” welches wichtige “alltägliche” und allgemeine Arbeit in den Zellen ausführt. Nachdem man die genetische Grundlage der SMA im Detail geklärt hatte, ist es nun Aufgabe der Biochemiker und Neurobiologen herauszufinden, welche Funktionen SMN in der Zelle ausführt, bzw. wie genau es bei Patienten zur Degeneration speziell der Motoneuronen kommt.
Ein entscheidender Schritt in diese Richtung war die Entwicklung einer Methode zur Isolierung des SMN-Proteins zusammen mit seinen Interaktionspartnern aus Zellen. Dies gelang unter Zuhilfenahme von spezifischen Antikörpern (das sind Eiweiße des Immunsystems), die das SMN-Protein binden. Studien mit diesen Antikörpern machten schnell klar, dass SMN in der Zelle in einem großen Proteinverband (“SMN-Komplex”) vorliegt, der mindestens noch 14 weitere Faktoren enthält. Glücklicherweise fanden sich unter den SMN-Interaktoren schon einige bekannte Proteine. Es handelt sich um die sogenannten Sm-Proteine, die wichtige Bestandteile einer der komplexesten zellulären Maschinerien überhaupt sind: dem Spleißosom. Das Spleißosom ist ein Gebilde aus Nukleinsäuren (RNA) und ca. 150 Proteinen, das der Zelle dabei hilft, seine Erbinformation in eine “lesbare” Form zu bringen. „Lesbar“ bedeutet, dass die Erbinformation abgelesen wird und daraus in zwei Schritten ein funktionsfähiger Eiweißbaustein (Protein) entsteht. Die Erbinformation ist jedoch nicht wie Sätze in einem Buch lückenlos und verständlich zusammengesetzt, sondern wird durch unsinnige Leseabschnitte unterbrochen. Diese werden beim Umschreiben der Erbinformation in die Boten-RNA (mRNA), dem ersten Schritt, zunächst übernommen, dann aber herausgeschnitten, damit im zweiten Schritt aus ihr nur ein funktionsfähiges und nicht fehlerhaftes Protein gebildet werden kann. Das Herausschneiden der unsinnigen Leseabschnitte übernimmt das Spleißosom, womit es eine zentrale Funktion bei der Umsetzung der genetischen Information einnimmt. Welche Funktion haben nun aber Proteine des Spleißosoms im SMN-Komplex? Hierüber gaben Untersuchungen an Fröschen Auskunft. Die unbefruchteten Eizellen der weiblichen Frösche (Oocyten) sind ein beliebtes Forschungsobjekt, da man aufgrund ihrer Größe zelluläre Prozesse sehr gut untersuchen kann. Mit Hilfe der Oocyten ließ sich feststellen, dass der SMN-Komplex dabei behilflich ist, Untereinheiten des Spleißosoms zusammenzubauen. Er erfüllt daher, bildlich gesprochen, die Funktion eines Bauarbeiters, der mehrere Komponenten (in diesem Fall Proteine und RNA) zu einer funktionsfähigen Einheit zusammenfügt (s. Abbildung). Die Existenz eines solchen „Baumeisters“ war für die Wissenschaftler eine große Überraschung, da man bislang dachte, dass derartige Prozesse in der Zelle spontan, d.h. ohne weitere Hilfe ablaufen.
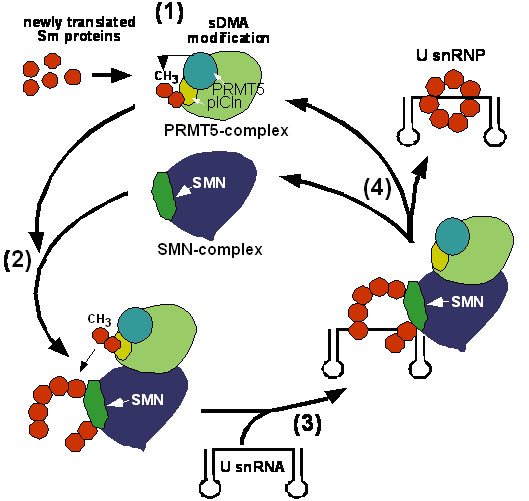
Im nächsten Schritt muss nun geklärt werden, wie der SMN-Komplex diese Funktion vermittelt. Konkret heißt das, dass wir verstehen möchten, welche Anteile des SMN-Komplexes bestimmte (Teil)-Prozesse der Zusammenlagerungs-Reaktion des Spleißosoms ausführen und welches die treibenden Kräfte hierbei sind. Erste Ergebnisse wurden auch hier schon erzielt. So konnte man feststellen, dass die Funktion des SMN-Komplexes durch einen weiteren Proteinverband (PRMT5-Komplex genannt) streng kontrolliert und reguliert wird. Dieser Komplex hängt chemische Seitengruppen (Methylgruppen) an die Sm-Proteine und aktiviert sie somit für die Zusammenlagerung (s. Abbildung).
Aufgrund der oben zusammengefassten Daten, die man zumeist erst kürzlich erhalten hat, ist klar geworden, dass SMN in ein völlig neuartiges Proteinnetzwerk integriert ist, welches eine bis vor kurzem unbekannte zelluläre Funktion (Zusammenlagerung von Spleißosomen-Untereinheiten) ausführt. Es stellt sich aber immer wieder die Frage, ob dies die alleinige Funktion von SMN ist, oder ob es noch andere Aktivitäten gibt, die durch diesen Faktor vermittelt werden. Dies wäre nichts ungewöhnliches, weiß man doch, dass viele Proteine mehrere Funktionen innerhalb der Zelle ausüben können. In der Tat hat man bei der Analyse von SMN in Motorneuronen eine Entdeckung gemacht, die auf eine weitere Funktion hinweist: Hier befindet sich das Protein in den langen Fortsätzen („Axons“) der Neuronen, die den Muskel zur Kontraktion reizen. Diese und weitere Untersuchungen lassen daher vermuten, dass SMN auch an axonalen Prozessen wie z.B. dem Transport von zellulären Komponenten durch das Axon, beteiligt ist.
Doch zurück zu der entscheidenden Frage. Wie kommt es zum Krankheitsbild der SMA? Mit anderen Worten: welcher Pathomechanismus führt in den Motorneuronen von SMA-Patienten zu deren fataler Degeneration? Bevor man sich dieser Frage zuwendet, muss man sich nochmals vergegenwärtigen, wie die SMA hervorgerufen wird: Es ist die Reduktion der SMN-Proteinmenge und nicht der vollständige Verlust, der die Motorneuronen-Degeneration verursacht. Der völlige Verlust der SMN-Expression ist nicht mit dem Leben vereinbar und führt, wie in einem Maus-Krankheitsmodell für die SMA gezeigt, zum frühzeitigen Absterben der Embryos.
Es sind aufgrund theoretischer Überlegungen mindestens zwei Hypothesen zur Entstehung der SMA denkbar, die von verschiedenen Forschern vertreten und untersucht werden. Einerseits könnte die Reduktion der SMN-Produktion zu einem Defekt bei der Zusammenlagerung des Spleißosoms führen. Dies führt entweder direkt oder indirekt über Sekundäreffekte, wie z.B. die verminderte oder fehlerhafte mRNA Produktion, zum Untergang der Motorneuronen.
Hier stellt sich unmittelbar die Frage, warum ein Defekt in einem solchen allgemeinen Prozess (alle Zellen brauchen das Spleißosom für die Expression ihrer Gene) nur die Motorneuronen, sonst aber keine anderen Zelltypen trifft. Die mögliche Antwort darauf könnte sein, dass einige Zelltypen, insbesondere die Motoneuronen, größere Mengen an Spleißosomen benötigen als andere und daher leichter untergehen.
Andererseits lässt sich postulieren, dass SMN noch eine weitere Funktion hat, die Motorneuronen-spezifisch wäre, wie z.B. den oben angesprochenen axonalen Transport bestimmter Komponenten. Durch die reduzierte Aktivität von SMN in diesem Prozess würden somit nur die Motorneuronen betroffen sein, da dieser Transportprozess ja nur hier auftritt. Hierbei müsste man allerdings ergänzend postulieren, dass die generelle Funktion von SMN beim Spleißosomaufbau bei den Patienten dann nicht so weit gestört sein darf, dass sie schon zuvor zur Degeneration der Neurone führt.
Die SMA ist heute eine der auf molekularer Ebene am besten verstandenen neuromuskulären Krankheiten überhaupt. Dies liegt nicht zuletzt an der äußerst effizienten und stimulierenden Zusammenarbeit von Forschern aus unterschiedlichen Fachrichtungen, einschließlich der Medizin, Genetik, Biochemie und Neurobiologie. Dieser Initiative ist es auch zu verdanken, dass nun schon seit einiger Zeit Versuche unternommen werden, die Krankheit medikamentös zu behandeln. In den meisten Fällen zielen Therapieansätze darauf ab, die Expression von SMN im Körper zu steigern, um so den zellulären Defekt (welcher es auch immer sein mag) aufzuheben. Es bleibt zu hoffen, dass diese hoffnungsvollen Ansätze schon bald eine Behandlung der SMA ermöglichen.
Prof. Dr. Utz Fischer
Biozentrum der Universität Würzburg